流行性感冒病毒核酸检测试剂主要原材料研究资料、主要生产工艺及反应体系的研究资料指导原则的通知
一,主要原材料研究资料
应提供主要原材料如引物、探针、企业参考品或标准品的选择与来源、制备过程、质量分析和质控标准等的相关研究资料。若主要原材料为企业自己生产,其生产工艺必须相对稳定;如主要原材料购自其他供货商,应提供的资料包括:对物料供应商审核的相关资料、供货方提供的质量标准、出厂检定报告,以及该原材料到货后的质量检验资料。
1.核酸分离/纯化组分(如有)的主要组成、原理介绍及相关的验证资料。
2.RT-PCR组分的主要材料(包括引物、探针、各种酶及其他主要原料)的选择、制备、质量标准及实验研究资料,主要包括以下内容:
(1)脱氧三磷酸核苷(dNTP)
核酸的组成成分,包括:dATP、dUTP、dGTP、dCTP和dTTP,对纯度、浓度、保存稳定性等验证资料。
(2)引物
由一定数量的dNTP构成的特定序列,通常采用DNA合成仪人工合成,合成后经聚丙烯酰胺凝胶电泳(PAGE)或其他适宜方法纯化。需提供对序列准确性、纯度、稳定性、功能性实验等验证资料。如为外购,应提供合成机构出具的合成产物的质检证明,如PAGE电泳结果或高效液相色谱法(HPLC)分析图谱。
(3)探针
特定的带有示踪物(标记物)的已知核酸片段(寡聚核苷酸片段),能与互补核酸序列退火杂交,用于特定核酸序列的探测。合成后经聚丙烯酰胺凝胶电泳或其他适宜方法纯化,在5-端(和/或3-端)进行标记,并经HPLC或其他适宜方法纯化。纯度应达到HPLC纯,应提供合成机构出具的合成产物的质检证明。
(4)PCR反应所需酶
DNA聚合酶,应具有DNA聚合酶活性,无核酸内切酶活性,具热稳定性,如:94℃保温1小时后仍保持50%活性;尿嘧啶糖基化酶(UNG),具有尿嘧啶糖基化活性,无核酸外切酶及核酸内切酶活性,应对酶活性有合理验证;逆转录酶,具逆转录酶活性,无核酸内切酶活性。应提供有关保存稳定性、活性及功能实验等的验证资料。
3.对照品(质控品)的原料选择、制备、定值过程及试验资料。
4.核酸类检测试剂的包装材料和耗材应无DNase和RNase污染。
二,主要生产工艺及反应体系的研究资料
基本生产工艺主要包括:配制工作液、半成品检定、分装和包装。配制工作液的各种原材料及其配比应符合要求,原材料应混合均匀,配制过程应对pH、电导率等关键参数进行有效控制。
生产工艺研究资料应能对反应体系涉及到的基本内容,如临床样本用量、试剂用量、反应条件、质控体系设置、Ct(临界)值确定等,提供确切的依据,主要包括以下内容:
1.主要生产工艺介绍,可以图表方式表示。
2.反应原理介绍。
3.基因位点选择、RT-PCR方法学特性介绍。
4.确定最佳RT-PCR反应体系的研究资料,包括酶浓度、引物/探针浓度、dNTP浓度、阳离子浓度等。
5.确定RT-PCR反应各阶段温度、时间及循环数的研究资料。
6.对于基线阈值(threshold)和阈值循环数(Ct)确定的研究资料。
7.不同适用机型的反应条件如果有差异应分别详述。
另外,对于试剂盒的对照(质控)品设置,建议企业参考以下要求执行:
(1)流感病毒核酸检测试剂盒的外部对照(质控)品应至少设置临界阳性对照(质控)品和阴性对照(质控)品,均应参与样本核酸的平行提取,以对核酸提取、RT-PCR反应过程、试剂/设备、交叉污染等环节进行合理质量控制,企业应对各种对照(质控)品的Ct值做出明确的范围要求。注意,建议采用与实际检测样本具有相同或相似性状的基质溶液作为阴性对照(质控)品,不推荐采用水作为阴性对照(质控)品。
(2)样本反应管应设置合理的内对照(内标)以对管内抑制可能造成的假阴性结果进行质控。申请人应对内标的引物、探针和模板的浓度做精确验证,既要保证内标荧光通道呈明显的阳性曲线又要尽量降低对靶基因检测造成的竞争性抑制而导致假阴性。对内标的Ct值也应有明确的范围要求。
(3)关于对照品的原料选择:内对照(内标)应采用具有蛋白外壳的病毒颗粒,如灭活的流感病毒或缺陷病毒(假病毒)等,外部阳性对照可以采用灭活病毒、假病毒或质粒。
表1 建议用于交叉反应性研究的微生物

*项:选择性验证。
(2)干扰物质
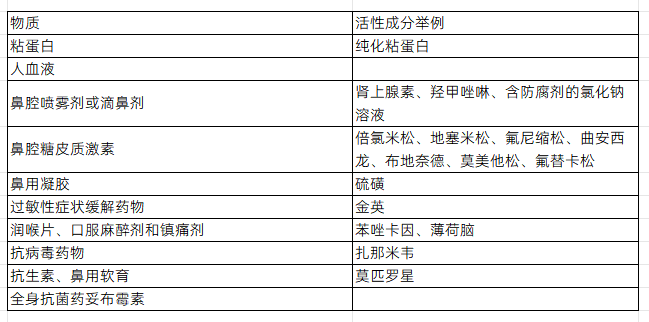
①潜在的干扰物质主要包括:血液、鼻分泌物或粘液、用于缓解鼻塞和咽部充血、鼻腔干燥、刺激、哮喘和过敏症状的药物(见表2)。
②使用医学相关水平的干扰物浓度进行验证,另外,建议申请人在每种干扰物质的潜在最大浓度(“最差条件”)条件下进行评价。
③申请人应采用每种流感病毒亚型的至少两种病毒株对呼吸道样本中物质的潜在抑制影响进行评估,建议在每种流感病毒的检测临界值水平对每种干扰物质的干扰影响进行检测。
表2 建议用于干扰研究的物质
4.精密度
测量精密度的评价方法并无统一的标准可依,可根据不同产品特征或企业的研究习惯进行,前提是必须保证研究的科学合理性。具体实验方法可以参考相关的CLSI-EP文件或国内有关体外诊断产品性能评估的文件进行。企业应对每项精密度指标的评价标准做出合理要求,如标准差或变异系数的范围等。针对本类产品的精密度评价主要包括以下要求。
(1)对可能影响检测精密度的主要变量进行验证,除申报试剂(包括提取组分和RT-PCR组分)本身的影响外,还应对PCR分析仪、操作者、地点等要素进行相关的验证。
(2)合理的精密度评价周期,例如:为期至少20天的连续检测,每天至少由2人完成不少于2次的完整检测,从而对批内/批间、日内/日间以及不同操作者之间的精密度进行综合评价。如有条件,申请人应选择不同的实验室进行重复实验以对室间精密度进行评价。
(3)用于精密度评价的质控品应至少包括3个水平:
①阴性质控品:待测物浓度低于最低检测限或为零浓度,阴性检出率应为100%(n≥20)。
②临界阳性质控品:待测物浓度略高于试剂盒的最低检测限,阳性检出率应高于95%(n≥20)。
③阳性质控品:待测物浓度呈中度到强阳性,阳性检出率为100%且CV≤10%(n≥20)。
另外,建议申请人选择适量临床采集的新鲜病人样本(包括所有样本类型)作为无靶值质控品进行精密度评价,以更好地模仿临床检测环境。
5.阳性/阴性参考品
如申报产品有相应的国家参考品,则企业内部阳性/阴性参考品应参考国家参考品的项目设置。在不低于国家参考品要求的前提下,申请人可以结合实际情况设置合理的企业内部阳性/阴性参考品。对于没有国家参考品的产品,申请人应根据产品性能验证的实际情况自行设定企业内部参考品,阳性参考品应着重考虑病毒型别、亚型及滴度要求,阴性参考品则主要涉及对分析特异性(交叉反应)的验证情况。
申请人应对内部阳性/阴性参考品的来源、型别鉴定、病毒滴度等信息进行精确的实验验证,并提交详细的验证资料。
6.其他需注意问题
对于适用多个机型的产品,应提供如产品说明书【适用机型】项中所列的所有型号仪器的性能评估资料。